Skeletal muscle insulin resistance in type 2 diabetes is associated with a shift from oxidative to glycolytic metabolism in myofibers. However, whether this metabolic switch is detrimental or adaptive for metabolic homeostasis has not been resolved. We recently demonstrated that the Baf60c/Deptor pathway promotes glycolytic metabolism in the muscle and protects mice from diet-induced insulin resistance. However, the nature of the signals that impinge on this pathway and the role of Baf60c in glucose homeostasis in the severe insulin-resistant state remain unknown. Here we show that expression of Baf60c and Deptor was downregulated in skeletal muscle in obesity, accompanied by extracellular signal–related kinase (ERK) activation. In cultured myotubes, inhibition of ERK, but not Jun NH2-terminal kinase and IκB kinase, blocked the downregulation of Baf60c and Deptor by the proinflammatory cytokine tumor necrosis factor-α. Treatment of obese mice with the ERK inhibitor U0126 rescued Baf60c and Deptor expression in skeletal muscle and lowered blood glucose. Transgenic rescue of Baf60c in skeletal muscle restored Deptor expression and Akt phosphorylation and ameliorated insulin resistance in ob/ob mice. This study identifies the Baf60c/Deptor pathway as a target of proinflammatory signaling in skeletal muscle that may link meta-inflammation to skeletal myofiber metabolism and insulin resistance.
Introduction
Skeletal muscle insulin resistance in patients with type 2 diabetes has been associated with impaired mitochondrial oxidative phosphorylation (OXPHOS) (1–3) and a shift from oxidative to glycolytic myofiber types (4,5). These observations led to the hypothesis that reduced oxidative metabolism in skeletal myofibers may be causally linked to the pathogenesis of muscle insulin resistance. Paradoxically, muscle-specific transgenic expression of peroxisome proliferator–activated receptor (PPAR)-γ coactivator 1α (PGC-1α), a transcriptional coactivator that promotes oxidative myofiber formation (6), improves insulin sensitivity in aged mice while exacerbating high-fat diet (HFD)-induced skeletal muscle insulin resistance (7,8). Recent clinical studies demonstrated that resistance training, which promotes the growth and function of fast-twitch glycolytic muscles, improves metabolic profiles in diabetic patients (9,10). Further, inhibition of myostatin (Mstn) signaling and conditional Akt1 activation in skeletal muscle improve the metabolic profile in diet-induced obesity through promoting glycolytic muscle metabolism in mice (11,12). As such, the cause-and-effect relationship between myofiber metabolism and insulin sensitivity remains uncertain.
The expression of myosin heavy-chain (MHC) isoforms is used to classify skeletal muscle fibers as fast-twitch or slow-twitch fibers (13). In general, slow-twitch fibers contain high mitochondrial content and rely predominantly on oxidative metabolism, whereas fast-twitch fibers are more glycolytic. At the molecular level, PGC-1α and its transcriptional partners have emerged as key regulators of oxidative metabolism in skeletal myofibers (6,13,14). We recently identified Baf60c, a subunit of the SWI/SNF chromatin-remodeling complex that interacts with selective transcription factors, as a core component of a regulatory cascade that drives glycolytic myofiber formation (15). Muscle-specific transgenic expression of Baf60c within physiological range promotes a shift of metabolic and contractile function toward the glycolytic fiber type through Deptor-mediated Akt activation. Interestingly, transgenic activation of the glycolytic muscle program by Baf60c protects mice from diet-induced insulin resistance and glucose intolerance. This work illustrates that the oxidative to glycolytic metabolic shift in skeletal muscle is potentially adaptive and beneficial in metabolic stress conditions.
Chronic and low-grade inflammation, or meta-inflammation, has been implicated as a major factor in obesity-associated insulin resistance (16–18). The molecular and cellular mechanisms linking meta-inflammation to hepatic and adipose tissue dysfunction and insulin resistance have been extensively documented (19–22). Skeletal muscle secretes diverse cytokines and also serves as a target of cytokine signaling (23). Notably, plasma levels of tumor necrosis factor-α (TNF-α), interleukin 6 (IL6), and Mstn are elevated in obese rodents and humans, potentially contributing to muscle insulin resistance (24–26). Beyond local actions in skeletal muscle, IL6 is known to exert complex metabolic effects on other peripheral tissues, such as adipose tissues and the liver, and may exert beneficial effects on systemic metabolism (27,28). Here we show that the Baf60c/Deptor pathway is downregulated in skeletal muscle during obesity through an extracellular signal–related kinase (ERK)-dependent mechanism. Transgenic rescue of Baf60c in skeletal muscle promotes glycolytic metabolism and ameliorates glucose intolerance and insulin resistance in ob/ob mice. This study identified the Baf60c/Deptor pathway as a target of proinflammatory signaling in skeletal muscle that may link meta-inflammation to skeletal myofiber metabolism and insulin resistance.
Research Design and Methods
Animal Studies
Generation of muscle-specific Flag/HA-tagged Baf60c (MCK-FH-Baf60c) mice has been described previously (15). The ob/ob and db/db mouse strains were purchased from The Jackson Laboratory, and ob/+ mice were crossed with MCK-FH-Baf60c mice to generate ob/ob MCK-FH-Baf60c mice. The comparison was performed between ob/ob MCK-FH-Baf60c (ob/ob transgenic [Tg]) mice and their littermate ob/ob controls. The muscle-specific coxsackie adenovirus receptor (MCK-CAR) transgenic strain was a gift from Drs. Nalbantoglu and Holland (McGill University). All animal studies were performed according to procedures approved by the University of Michigan Committee on Use and Care of Animals.
Adenoviral Transduction and Histological Analysis
Adenoviruses expressing control short hairpin (sh)RNA or Deptor shRNA were purified and injected intramuscularly into transversus abdominis (TA) muscle of MCK-CAR mice, as previously described (15). Muscle samples were immediately frozen in isopentane, precooled with liquid nitrogen after dissection, and sectioned on a cryostat-microtome. Histochemical staining for succinate dehydrogenase (SDH), α-glycerol-6-phosphate dehydrogenase (α-GPDH), and cytochrome c oxidase (COX) enzymes was performed as described (15).
Insulin Tolerance Test and Glucose Tolerance Test Studies
For the glucose tolerance test (GTT), a glucose solution in saline was injected intraperitoneally into mice fasted overnight at 2.0 g/kg body weight. Immediately before, and 15, 30, 60, and 120 min after the glucose injection, blood glucose levels were measured by tail bleeding. For the insulin tolerance test (ITT), mice were starved for 4 h and intraperitoneally injected with insulin solution in saline at a dose of 2.8 units/kg body weight. Blood glucose concentrations were measured before and 20, 45, 90, and 120 min after the insulin injection.
Muscle Cell Culture and Differentiation
C2C12 myoblasts were obtained from American Type Culture Collection and maintained in Dulbecco’s modified Eagle’s medium containing 10% FBS. Pooled primary human skeletal myoblasts isolated from three healthy adult donors were purchased from Zen-Bio (Research Triangle Park, NC) and cultured in skeletal muscle growth medium (SKM-M, Zen-Bio). All experiments in this study were conducted on differentiated myotubes unless otherwise indicated. To induce myotube differentiation, growth media were replaced with Dulbecco’s modified Eagle’s medium containing 2% bovine growth serum or skeletal muscle differentiation medium (SKM-D, Zen-Bio) for confluent C2C12 or primary human myoblasts, respectively.
Molecular Analyses
Quantitative PCR (qPCR), immunoblotting, and chromatin immunoprecipitation (ChIP) assay were performed as previously described (15). For the ChIP assay, C2C12 or primary human myotubes were fixed with 1% formaldehyde and sonicated to produce chromatin lysates. The lysates were precleared with Protein-G agarose beads and immunoprecipitated overnight with antibodies against Ace-H3 (Millipore), Ace-H4 (Millipore), H3K4m3 (Millipore), H3K9m2 (Abcam), or control IgG in the presence of BSA and salmon sperm DNA. The next day, Protein-G agarose beads were added to each immunoprecipitation reaction for 1 h, followed by an extensive wash and reverse crosslink. DNA was eluted from the beads and purified using a PCR Purification kit (Invitrogen) and subsequently analyzed by qPCR using primers located on the proximal Baf60c and Deptor promoters. Primers are available upon request.
Measurement of Lactate Dehydrogenase Activity
Skeletal muscle lactate dehydrogenase (LDH) activity was measured using an assay kit (BioVision). Briefly, frozen TA muscle was homogenized in 0.25 mL cold assay buffer. The supernatant was harvested after centrifugation at 10,000g for 15 min at 4°C to measure the LDH enzyme activity. Protein concentrations were determined by the Bradford method. LDH activity in the muscle lysate was normalized to the total protein amount and expressed as nmol/min/mg protein.
Glycolytic Flux and Fatty Acid Oxidation Assays
Soleus and plantaris muscles were dissected and used for glycolytic flux measurement and fatty acid oxidation assay using d-[5-3H]glucose and [1-14C]palmitate as substrates, respectively. 3H2O produced by glycolysis and 14CO2 generated in complete fatty acid oxidation were separated and measured by liquid scintillation counting, as previously described (15,29). Briefly, for the glycolytic flux assay, soleus and plantaris muscles dissected from ob/ob and ob/ob Tg male mice were incubated for 1 h at 37°C in Krebs buffer containing 10 μCi/mL d-[5-3H]glucose and 5 mmol/L glucose. 3H2O produced by glycolysis was separated from d-[5-3H]glucose in the incubation buffer using the equilibration method and determined by liquid scintillation counting. For the fatty acid oxidation assay, soleus and plantaris muscles were dissected and incubated for 2 h at 37°C in 0.5 mL of a modified Krebs-Ringer bicarbonate buffer (low calcium) containing 0.2 mmol/L nonradioactive palmitate, 1.0 µCi/mL [1-14C]palmitate, 1.0 mmol/L l-carnitine, 5 mmol/L glucose, and 0.5% BSA. 14CO2 generated by complete fatty acid oxidation was trapped in 200 µL sodium hydroxide (1N) and measured by liquid scintillation counting.
Statistics
Data were analyzed using the unpaired Student t test for independent groups. A P value of <0.05 was considered statistically significant.
Results
The Baf60c/Deptor Pathway Is Downregulated in Skeletal Muscle in Obesity
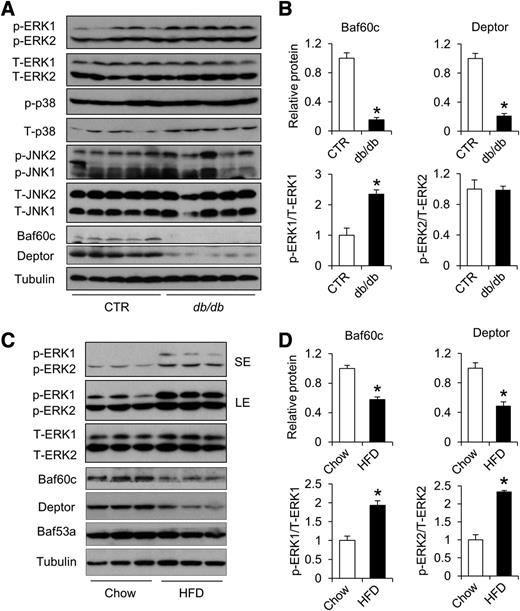
We recently demonstrated that Baf60c is enriched in fast-twitch muscles and promotes glycolytic metabolism in skeletal myofibers (15). Muscle-specific transgenic expression of Baf60c stimulates the expression of Deptor and protects the mice from HFD-induced insulin resistance. To assess whether the Baf60c/Deptor pathway is dysregulated in obesity, we first examined their expression in skeletal muscle from leptin-deficient ob/ob mice. Compared with lean control mice, mRNA levels of Baf60c and Deptor were significantly lower in the quadriceps from ob/ob mice (Fig. 1A). In contrast, mRNA expression of BAF60a and BAF60b, two other members of the BAF60 family, remained similar between the two groups. Immunoblotting analyses demonstrated that Baf60c and Deptor protein expression was reduced by ∼83 and 58%, respectively, in muscles from ob/ob mice (Fig. 1B and C).
Persistent and low-grade inflammation plays an important role in obesity-associated insulin resistance. The mitogen-activated protein kinase (MAPK) and nuclear factor (NF)-κB pathways have been implicated in linking meta-inflammation to insulin resistance in white adipose tissue (WAT) and the liver (19–22). However, the pathways that mediate the deleterious effects of chronic inflammation on skeletal muscle energy metabolism remain poorly defined. In mammals, the NF-κB transcriptional factor family consists of five members: RelA (p65), RelB, c-Rel, and the precursor proteins NF-κB1 (p105) and NF-κB2 (p100) (30). Activation of NF-κB pathways is initiated through phosphorylation of the inhibitory IκB protein by IκB kinases (IKKs), including IKK-α, IKK-β, IKK-ε, and TANK-binding kinase 1 (TBK1). The MAPK cascade contains three parallel arms, the ERK1/2, Jun NH2-terminal kinase (JNK) 1/2, and p38 pathways (31).
We next performed immunoblotting assays using total protein lysates from the quadriceps muscle to determine whether these modules are altered in obesity. As shown in Fig. 1D, phosphorylated and total protein levels of TBK1, IKKε, IκBα, p105, and p65, were similar in muscles from wild-type and ob/ob mice. Protein levels of p38 and JNK1 were also similar (Fig. 1E and F), whereas JNK2 phosphorylation was slightly increased compared with control mice. In contrast, phosphorylation of ERK1 and ERK2 was significantly enhanced in quadriceps from ob/ob mice, whereas total ERK1/2 expression remained similar. Reduced Baf60c and Deptor expression associated with ERK activation was also observed in skeletal muscles from leptin receptor-deficient db/db (Fig. 2A and B) and diet-induced obese mice (Fig. 2C and D).
TNF-α Represses the Expression of Baf60c and Deptor in Cultured Mouse and Human Muscle Cells
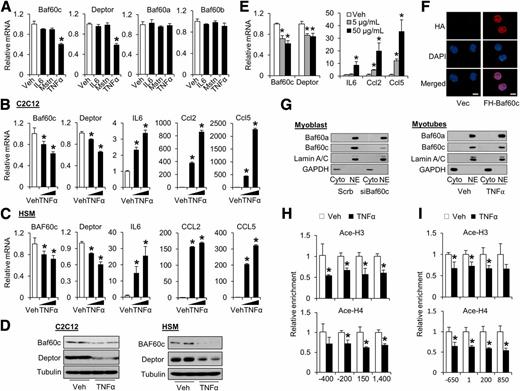
Previous studies have identified cytokines IL6, Mstn, and TNF-α as important mediators of skeletal muscle insulin resistance in obesity (24–26). To determine whether the Baf60c/Deptor pathway is a target of these cytokines, we treated C2C12 myotubes with IL6, Mstn, or TNF-α and performed gene expression analysis. As expected, TNF-α stimulated IL6, Ccl2, and Ccl5 mRNA expression. TNF-α, but not Mstn and IL6, significantly decreased Baf60c and Deptor mRNA expression in a dose-dependent manner (Fig. 3A and B). Similar inhibition of BAF60c and Deptor expression in response to TNF-α treatment was observed in cultured primary human myotubes (Fig. 3C). Consistently, Baf60c and Deptor protein levels were also reduced by TNF-α treatments in C2C12 and primary human myotubes (Fig. 3D). The inhibitory effects of TNF-α on Baf60c and Deptor expression were also observed in vivo after intramuscular injection of TNF-α (Fig. 3E). Baf60c was recently shown to undergo nuclear translocation in response to insulin in hepatocytes (32). To examine subcellular localization of Baf60c in muscle cells, we performed immunofluorescence and fractionation studies. As shown in Fig. 3F and G, Baf60a and Baf60c were exclusively detected in the nucleus independent of TNF-α treatments.
Baf60c is a subunit of the SWI/SNF chromatin remodeling complex, serving as a link between DNA-binding transcriptional factors and the core SWI/SNF complex (33). Changes in local chromatin structure and histone modifications play a critical role in the control of gene transcription (34). To explore whether TNF-α represses Baf60c and Deptor expression through epigenetic mechanisms, we performed ChIP assays in primary human myotubes treated with vehicle or TNF-α. As shown in Fig. 3H, TNF-α treatment markedly reduced the levels of acetyl-histone H3 (Ace-H3) and -histone H4 (Ace-H4), histone markers associated with transcriptionally active chromatin, in the proximal BAF60c promoter. Similarly, the levels of Ace-H3 and Ace-H4 in the proximal Deptor promoter were also significantly decreased by TNF-α treatment (Fig. 3I). In contrast, dimethylation of H3 lysine 9 (H3K9me2), a hallmark of transcriptional repression, was augmented in response to TNF-α in the proximal Deptor promoter (Supplementary Fig. 1A). Further, ChIP assays using TNF-α–treated C2C12 myotubes revealed that TNF-α treatment resulted in a robust shift of chromatin markers from a transcriptionally active to repressive state within the Deptor locus (Supplementary Fig. 1B). These results suggest that TNF-α downregulates the expression of Baf60c and Deptor through epigenetic mechanisms.
ERK Activation Mediates the Downregulation of Baf60c and Deptor Expression in Skeletal Muscle During Obesity
As shown above, obesity-induced downregulation of Baf60c and Deptor in skeletal muscle was associated with increased ERK1/2 phosphorylation, but to a lesser extent, changes in the JNK and NF-κB pathways. To assess the significance of these pathways in mediating the downregulation of Baf60c and Deptor, we treated differentiated C2C12 myotubes with TNF-α in the absence or presence of specific inhibitors. Immunoblotting analyses indicated that TNF-α treatment robustly stimulated ERK and JNK phosphorylation (Fig. 4A). As expected, preincubation of myotubes with PD98059, an ERK-specific inhibitor, or SP600125, a JNK-specific inhibitor, abolished TNF-α–induced phosphorylation of ERK1/2 and JNK1/2, respectively, whereas pretreatment with compound VIII, an IKK-β inhibitor, completely reversed TNF-α–induced phosphorylation of p105 and degradation of IκBα (Fig. 4B). Interestingly, PD98059 treatment reversed the inhibitory effects of TNF-α on the expression of Baf60c and Deptor in cultured myotubes (Fig. 4C). At a higher dose (50 μmol/L), PD98059 nearly completely rescued the downregulation of Baf60c and Deptor by TNF-α. On the contrary, preincubation with JNK inhibitor SP600125 had a modest effect on Baf60c and Deptor expression in the presence or absence of TNF-α (Fig. 4D). Similarly, compound VIII failed to relieve the inhibition of Baf60c and Deptor expression by TNF-α (Fig. 4E). Further, incubation of C2C12 myotubes with CAY10576 and Amlexanox, two inhibitors of noncanonical IKKs (35,36), had modest effects on mRNA expression of Baf60c and Deptor. Together, these results suggest that the downregulation of muscle Baf60c and Deptor expression in obesity may result from augmented inflammatory signaling through the ERK MAPK pathway.
Inhibition of the ERK pathway has been linked to improved glucose homeostasis (37,38). To directly assess the role of ERK activation in Baf60c/Deptor expression, we treated HFD-fed mice with vehicle or U0126, a specific ERK inhibitor, and analyzed skeletal muscle gene expression. As expected, 2 weeks of U0126 treatment (10 mg/kg) significantly decreased fasting blood glucose levels in diet-induced obese mice (Fig. 5A). Notably, inhibition of ERK1/2 phosphorylation restored mRNA and protein expression of Baf60c and Deptor in skeletal muscle (Fig. 5B–D). Similar effects of ERK inhibition were also observed in db/db mice (Fig. 5E–H). Taken together, these data demonstrate that obesity-associated inflammation attenuates Baf60c and Deptor expression, at least in part, through ERK activation in skeletal muscle.
Deptor Is Required for Maintaining Glycolytic Metabolism in Adult Skeletal Muscles
We recently showed that Deptor is a direct target of Baf60c that mediates its stimulation of Akt activation and glycolytic metabolism in cultured myotubes. However, the role of Deptor in muscle metabolism in adult mice has not been determined. Using MCK-CAR mice, we knocked down endogenous Deptor in TA muscle and examined muscle Akt activation and energy metabolism. Compared with contralateral control, Deptor shRNA significantly decreased endogenous Deptor mRNA and protein expression (Fig. 6A and B). Akt phosphorylation on both Thr-308 and Ser-473 residues was also attenuated by Deptor knockdown (Fig. 6B). Histochemical staining using substrate for glycolytic enzyme α-GPDH revealed that glycolytic metabolism was reduced after RNA interference knockdown of Deptor (Fig. 6C). In contrast, the activities of SDH and COX, enzymes involved in oxidative metabolism, were elevated in the transduced muscle expressing Deptor shRNA in comparison with control shRNA. Consistently, LDH activity was also significantly reduced by Deptor knockdown (Fig. 6D). We conclude from these data that Deptor is required for maintaining glycolytic metabolism in skeletal muscle in adult mice.
We previously demonstrated that Deptor cell-autonomously regulates the Akt pathway in cultured myotubes (15). To further examine the role of Deptor in insulin signaling and glucose utilization in muscle cells, we performed knockdown studies in cultured C2C12 myotubes. Compared with control, RNA interference knockdown of Deptor markedly reduced glucose utilization and lactate production in cultured myotubes (Fig. 6E). In addition, glycolytic flux measurements using d-[5-3H]glucose indicated that Deptor knockdown attenuated insulin-stimulated glycolytic flux in transduced myotubes (Fig. 6F). These data suggest that Deptor regulates muscle Akt activation and glycolytic metabolism in a cell-autonomous manner.
Muscle-Specific Transgenic Expression of Baf60c Promotes Glycolytic Metabolism and Improves Whole-Body Glucose Homeostasis in ob/ob Mice
We next examined whether transgenic expression of Baf60c in skeletal muscle is sufficient to improve glucose homeostasis in severe obesity. We crossed MCK-FH-Baf60c transgenic mice with ob/+ mice and examined skeletal muscle and systemic metabolism in ob/ob and ob/ob MCK-FH-Baf60c (ob/ob Tg) mice. qPCR analyses of skeletal muscle gene expression indicated that mRNA levels of several glycolytic enzymes, including glucose phosphate isomerase 1 (Gpi1), triosephosphate isomerase 1 (Tpi1), and phosphoglycerate mutase 2 (Pgam2), were significantly elevated in transgenic quadriceps muscles (Fig. 7A). In addition, the expression of fructose bisphosphatase 2 (Fbp2), which catalyzes the reverse reaction of the rate limiting glycolytic enzyme phosphofructose kinase (Pfkm), was lower in ob/ob Tg than ob/ob muscles. In contrast, mRNA and protein expression of several genes involved in lipid uptake (CD36 antigen [CD36], VLDL receptor [VLDLR]), fatty acid β-oxidation (carnitine palmitoyltransferase 1b, muscle [Cpt1β]; acyl-CoA dehydrogenase, medium chain [Acadm]; acyl-CoA dehydrogenase, long-chain [Acadl]; acyl-CoA dehydrogenase, very long chain [Acadvl]), and mitochondrial OXPHOS (NADH dehydrogenase [ubiquinone] 1 β subcomplex 8 [Ndufb8]; succinate dehydrogenase complex, subunit B [Sdhb]; ubiquinol cytochrome c reductase core protein 2 [Uqcrc2]; mitochondrially encoded COX I [Mtco1]; ATP synthase, H+ transporting, mitochondrial F1 complex, α subunit 1 [Atp5a1]; and NADH dehydrogenase [ubiquinone] flavoprotein 2 [Ndufv2]) was significantly lower in transgenic muscle. Consistent with our previous studies, Baf60c transgenic expression increased mRNA and protein expression of Deptor (Fig. 7B and C). Further, SDH enzymatic activity was decreased, whereas α-GPDH activity was increased, in transgenic TA and extensor digitorum longus muscles (Fig. 7D). Measurements of fatty acid oxidation rate and glycolytic flux in isolated muscles ex vivo revealed that Baf60c transgenic expression significantly enhanced glycolytic flux, while having modest effect on fatty acid oxidation (Fig. 7E and F). These results demonstrate that transgenic expression of Baf60c is sufficient to promote a molecular and functional program of glycolytic metabolism in skeletal muscle from ob/ob mice.
Compared with ob/ob, ob/ob Tg mice had significantly lower fasting blood glucose levels (Fig. 8A). Plasma insulin concentrations trended lower. The GTT and ITT results revealed that, compared with control, ob/ob Tg mice had improved glucose tolerance and insulin sensitivity (Fig. 8B). Although plasma triglyceride and nonesterified fatty acid levels were similar between the two groups, the concentration of plasma ketone body was higher in the transgenic group (Fig. 8C). Baf60c exerts its effects on glycolytic metabolism through its induction of Deptor and stimulation of Akt phosphorylation. Immunoblotting analyses revealed that transgenic expression of Baf60c also augmented Akt phosphorylation in muscle from ob/ob mice (Fig. 8D and E). AMP-activated protein kinase phosphorylation was slightly increased in the transgenic group. In contrast, phosphorylation of S6 kinase 1 (S6K1) and S6 (Fig. 8D), hallmarks of mammalian target of rapamycin activity, and myokine gene expression (Fig. 8F) remained similar between the two groups despite increased Deptor expression.
We next performed histological and gene expression analyses in the liver, brown adipose tissue (BAT) and WAT from ob/ob and ob/ob Tg mice. Unlike MCK-Baf60c transgenic mice in C57BL/6J background, which are protected from diet-induced obesity and hepatic steatosis, ob/ob and ob/ob Tg mice had similar body weight, WAT-to-body weight and liver-to-body weight ratio, and liver triglyceride content (Supplementary Fig. 2A). The histological appearance of the liver, WAT, and BAT was nearly indistinguishable between the two groups (Supplementary Fig. 2B). Gene expression analyses revealed that the expression of major metabolic genes and transcriptional regulators in the liver and adipose tissues was largely unaffected by transgenic expression of Baf60c in the ob/ob background (Supplementary Fig. 2C). As such, the improvement of glucose homeostasis in ob/ob Tg mice is likely a direct consequence of enhanced glycolytic metabolism driven by Baf60c in skeletal muscle.
Discussion
The regulation of glycolytic metabolism in skeletal muscle and its role in glucose homeostasis remain unresolved. We recently demonstrated that transcriptional cofactor Baf60c is enriched in glycolytic muscles and promotes glycolytic metabolism in the muscle through its induction of Deptor expression and Akt activation. Surprisingly, muscle-specific transgenic expression of Baf60c protects mice from HFD-induced insulin resistance (15), suggesting that skeletal muscle glycolysis serves a protective role in whole-body glucose metabolism. The expression of Baf60c is downregulated in skeletal muscle in rodent models of obesity. However, the nature of the physiological signals that repress the Baf60c/Deptor pathway in obesity remains unknown. In addition, whether stimulation of glycolytic metabolism in the muscle improves glucose homeostasis in severe obesity has not been addressed.
In this study, we found that the expression of Baf60c and Deptor was reduced in the muscle from ob/ob mice. Studies in cultured mouse and primary human myotubes revealed that the proinflammatory cytokine TNF-α induced repressive epigenetic modifications at the proximal Baf60c and Deptor promoters, resulting in the downregulation of Baf60c and Deptor expression in muscle cells. Dissection of signaling pathways downstream of TNF-α indicates that activation of the ERK pathway mediates the inhibitory effect of cytokines on Baf60c and Deptor expression. Finally, transgenic expression of Baf60c in the ob/ob genetic background augments glycolytic metabolic activity in skeletal muscle and improves whole-body glucose homeostasis.
Meta-inflammation plays a pivotal role in the development of insulin resistance and metabolic dysfunction in type 2 diabetes. Pharmacological inhibition of inflammation provides beneficial effects on obesity-induced insulin resistance and glucose intolerance in rodents and in humans (36,39,40). Previous studies have identified the JNK and IKK/NF-κB pathways as key links between meta-inflammation and insulin resistance in WAT and the liver (20–22). In skeletal muscle, we found that ERK activation was markedly augmented in diet-induced and genetic obese mice, whereas the JNK, p38 MAPK, and IKK/NF-κB pathways remained unaffected, suggesting that distinct signaling pathways may be engaged by proinflammatory cytokines in a tissue-specific manner. In addition to inflammatory stimuli, ERK activation is also modulated by fatty acid uptake and metabolism in skeletal muscle (41). Remarkably, ERK inhibition restored the expression of Baf60c and Deptor in cultured myotubes and in vivo in obese mice. Although the ERK pathway have been extensively studied in the context of mitogenic signal transduction (31), this pathway can also be activated by inflammatory cytokines and contributes to the attenuation of insulin signaling (42). These findings strongly suggest that ERK activation plays a uniquely important role in mediating the reduction of skeletal muscle Baf60c and Deptor expression during obesity.
The Baf60 subunit of the SWI/SNF chromatin-remodeling complex provides a link between the core complex and transcription factors to direct specific gene regulation (33). Three related members, including Baf60a, Baf60b, and Baf60c, exhibit distinct patterns of tissue distribution and are likely uniquely required for diverse biological processes. Baf60a and Baf60c participate in the regulation of developmental processes such as Schwann cell differentiation (43) and cardiac and skeletal muscle development (44,45). We recently demonstrated that Baf60a forms a transcriptional complex with PPAR-α and PGC-1α and stimulates the expression of hepatic genes involved in peroxisomal and mitochondrial fatty acid β-oxidation (46,47). In skeletal muscle, Baf60c physically interacts with transcription factor Six4 and coordinately regulates the program of glycolytic metabolism (15). Interestingly, Baf60c undergoes phosphorylation in response to p38α kinase in myocytes and insulin signaling in hepatocytes (32,45), suggesting that the Baf60 subunit of the SWI/SNF complex is important in the regulation of glucose and lipid metabolism in response to hormonal signals. Previous studies demonstrated that p38γ MAPK regulates PGC-1α expression and skeletal muscle adaptation during exercise (48,49). Whether p38γ directly modulates Baf60c in this metabolic adaptation remains currently unknown.
The expression of Baf60c and Deptor is enriched in glycolytic muscles. These two factors form a cell-autonomous regulatory pathway that appears to be critical for maintaining glycolytic metabolism in adult skeletal muscle. We previously demonstrated that Baf60c is required and sufficient to promote muscle glycolytic metabolism (15). In this study, we found that Deptor is required for maintaining glycolytic metabolism in skeletal muscle in a cell-autonomous manner. In accordance, muscle-specific transgenic expression of Baf60c in ob/ob background induced Deptor expression and Akt phosphorylation and enhanced glycolytic metabolism. The expression of several genes involved in glycolysis was elevated, whereas those involved in mitochondrial fatty acid β-oxidative and OXPHOS were reduced in transgenic muscles. These changes in metabolic gene expression were consistent with increased glycolytic and decreased oxidative enzymatic activities, as revealed by histochemical staining. However, direct measurements of fatty acid and glucose metabolism in isolated muscles ex vivo revealed that transgenic expression of Baf60c significantly enhanced glycolytic flux without altering the rate of fatty acid oxidation. These results suggest that the improvement of whole-body glucose metabolism in Baf60c transgenic mice may be a direct consequence of enhanced glycolytic metabolism.
Reduced mitochondrial oxidative capacity and a shift from oxidative to glycolytic muscle fiber type have been associated with impaired skeletal muscle insulin action in patients with type 2 diabetes and in elderly individuals (1–5). As such, the current paradigm posits that reduced mitochondrial oxidative metabolism potentially plays a causative role in the pathogenesis of insulin resistance. An unexpected result from our studies is that transgenic activation of glycolytic muscle by Baf60c significantly improves whole-body glucose homeostasis and insulin sensitivity in diet-induced and genetic obesity. This improvement of the metabolic profile occurs despite reduced mitochondrial gene expression and oxidative activity in transgenic muscle. These apparently paradoxical findings, however, are similar to mice that have increased fast-twitch glycolytic muscle content as a result of conditional Akt activation or myostatin deficiency in skeletal muscle (11,12). Together, these studies illustrate that increased glycolytic metabolism in the muscle does not impair whole-body glucose metabolism and lead to insulin resistance. Instead, maintaining adequate glycolytic capacity in skeletal muscle is critical for protection from obesity-associated insulin resistance.
Article Information
Acknowledgments. The authors thank Deborah M. Muoio and Tim Kovers (Duke University) for their technical advice on the fatty acid oxidation assay in isolated skeletal muscles and are grateful to Qi Yu and Crystal Rui for technical assistance and to laboratory members for discussion.
Funding. This work was supported by a postdoctoral fellowship (12POST9600010) from the American Heart Association to Z.-X.M. and by National Institutes of Health grants DK-095151 and DK-077086 to J.D.L.
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Author Contributions. Z.-X.M. and J.D.L. conceived the project and designed the study. Z.-X.M., L.W., and Y.X. performed the experiments. Z.-X.M. and J.D.L. analyzed the data and wrote the manuscript. J.D.L. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.