


Glucagon-like peptide (GLP)-1 mimetics have been reported to cause hypoglycemia when combined with sulfonylureas. This study investigated the impact of tolbutamide on the glucose dependence of the GLP-1–mediated effects on insulin, glucagon, and somatostatin secretion in the in situ perfused rat pancreas. At 3 mmol/l glucose, GLP-1 alone did not augment insulin secretion, whereas tolbutamide alone caused a rapid increase in insulin secretion. However, when GLP-1 and tolbutamide were administered simultaneously, insulin secretion increased significantly to 43.7 ± 6.2 pmol/min (means ± SE), exceeding the sum of the responses to GLP-1 (2.0 ± 0.6 pmol/min; P = 0.019) and tolbutamide (11.3 ± 3.8; P = 0.005) alone by a factor of 3.3. At 11 mmol/l glucose, co-infusion of GLP-1 and tolbutamide augmented insulin secretion to 141.7 ± 10.3 vs. 115.36 ± 14.1 (GLP-1) and 42.5 ± 7.3 pmol/min (tolbutamide). Interestingly, increases in somatostatin secretion, both by glucose and GLP-1, were consistently paralleled by suppression of glucagon release. In conclusion, we demonstrate uncoupling of GLP-1 from its glucose dependence by tolbutamide. This uncoupling probably explains the tendency of GLP-1 to provoke hypoglycemia in combination with sulfonylureas. The results suggest that closure of ATP-sensitive K+ channels by glucose might be involved in the glucose dependence of GLP-1’s insulinotropic effect and that somatostatin acts as a paracrine regulator of glucagon release.
The incretin hormone glucagon-like peptide (GLP)-1 is a promising new therapeutic option for treatment of type 2 diabetes. It significantly ameliorates glucose homeostasis in diabetic patients by, among other effects, enhancing insulin and suppressing glucagon secretion (1,2). Whereas it is well established that GLP-1 directly stimulates insulin and somatostatin secretion via receptors expressed on β- and δ-cells, no such clear evidence exists for α-cells. Thus, in addition, an involvement of paracrine mediators in the intra-islet regulation of glucagon (as well as insulin and somatostatin) secretion has been intensely discussed (3–5). In contrast to the majority of insulin-releasing drugs, GLP-1 is not effective below a glucose threshold level of 4.3 mmol/l and, even when given in supra-physiological doses, does not provoke hypoglycemia in fasting healthy subjects (6,7). However, in clinical studies including diabetic patients receiving both exendin-4, a GLP-1 mimetic, and sulfonylurea medication, hypoglycemia occurred in up to 36% of patients (8–11).
The mechanisms underlying GLP-1 signaling (especially the role of ambient glucose concentrations) have been widely investigated (3,12). According to the at present generally accepted model for stimulus-secretion coupling in pancreatic β-cells, exposure of β-cells to elevated glucose levels (>7–8 mmol/l) initiates an intracellular cascade of metabolic events, which results in exocytosis of insulin-containing secretory granules. Briefly, after uptake into the β-cell by the GLUT2 transporter, glucose is subject to glycolysis, generating substrates for mitochondrial oxidative phosphorylation with subsequent synthesis of ATP. The increase in the ATP/ADP concentration ratio leads to inhibition of ATP-sensitive K+ (KATP) channels, membrane depolarization, and activation of voltage-dependent Ca2+ channels. The resulting increase in intracellular Ca2+ causes Ca2+-dependent exocytosis of insulin-containing granules. After binding to its guanine nucleotide binding protein–coupled receptor and stimulating cAMP production, GLP-1 affects this cascade via different signaling pathways at several points, amplifying the β-cell’s secretory response (13).
However, none of the GLP-1–mediated effects alone appears to be able to trigger insulin secretion. Instead, GLP-1 depends on the presence of adequate glucose levels.
Sulfonylureas, which are widely used in the treatment of type 2 diabetes, induce insulin secretion by directly binding to the SUR1 subunit of KATP channels, thereby causing their closure and initiating the signaling pathway described above (14–16). In contrast to GLP-1, sulfonylureas such as tolbutamide stimulate insulin secretion even at very low blood glucose concentrations and, therefore, can provoke severe hypoglycemia; however, their insulinotropic potency decreases with falling glucose levels (17).
Because GLP-1 is unable to induce significant insulin secretion under hypoglycemic conditions, we investigated the effect of tolbutamide on the glucose dependence of the GLP-1–mediated effects on insulin, glucagon, and somatostatin secretion. As a model, we used the in situ perfused rat pancreas, studied at different glucose concentrations. If closure of KATP channels is an essential molecular mechanism behind the glucose dependence of the insulinotropic actions of GLP-1, sulfonylurea pretreatment would be expected to uncouple it and allow for stimulation of insulin secretion at hypoglycemic levels. In addition, the high occurrence of hypoglycemia in diabetic patients treated with both GLP-1 and sulfonylurea compounds might be explained.
RESEARCH DESIGN AND METHODS
Animals.
Male Wistar rats (300–375 g, 10–12 weeks) were purchased from Charles River (Sulzfeld, Germany) (n = 13) more than 1 week before the experiments were performed and had free access to standard rodent food and water. Animals were housed two per cage and followed a 12:12-h light-dark cycle. All animal studies were carried out with permission in accordance with the guidelines of Danish legislation governing animal experimentation (1987) and the National Institutes of Health publication number 85-23 (revised 1985).
Isolated perfused rat pancreas.
Nonfasted rats were anesthetized with an intraperitoneal injection of pentobarbital (50 mg/kg; Royal Veterinary and Agriculture University, Frederiksberg, Denmark), and the pancreata were dissected and perfused in situ (18). Briefly, the pancreas was isolated from the vascularization of the abdominal organs by appropriately placed ligatures, except for the attached segment of the duodenum. The duodenum was sectioned and a drainage tube inserted distal to the pancreas. The spleen, stomach, large intestine, and residuals of the small intestine were removed. The rat was killed by removal of the heart, and the pancreas was perfused in a single pass system through both the celiac and the superior mesenteric arteries via a catheter inserted into the adjacent abdominal aorta. All other aortic branches were ligated. The venous effluent was collected for 1-min intervals via an obstructing cannula inserted into the portal vein and stored at −20°C until analysis. The flow rate was kept constant at 4 ml/min. The perfusion medium consisted of a modified Krebs-Ringer bicarbonate buffer containing, in addition, 0.1% human serum albumin (Behringwerke, Marburg, Germany), 5% Dextran T-70 (Pharmacia Biotech, Uppsala, Sweden), 3 mmol/l glucose, and 5 mmol/l pyruvate, fumarate, and glutamate each. The perfusion medium was gassed with a 95% O2/5% CO2 mixture to achieve pH 7.4 and maintained at 37°C during the entire experiment. Tolbutamide (Sigma Aldrich, Schnelldorf, Germany) was dissolved in DMSO and infused into the arterial line using a syringe pump to give a final perfusate concentration of 185 μmol/l (50 mg/l). The final concentration of DMSO in the perfusion medium did not exceed 0.1%. Synthetic GLP-1 (7–37) (a gift from Novo Nordisk, Bagsværd, Denmark) was dissolved in 0.9% NaCl containing 1% human serum albumin. Its authenticity was confirmed by amino acid analysis, analytical reversed-phase high-performance liquid chromatography, and plasma desorption mass spectrometry, and its purity was shown to be >99% by high-performance liquid chromatography with detection at 214 nm. The final concentration in the perfusate was 1 nmol/l. To raise the glucose concentration in the perfusion medium temporarily from 3 to 11 mmol/l, a concentrated glucose solution (500 g/l; SAD, Copenhagen, Denmark) was administered via the sidearm syringe according to the experimental protocol.
Experimental protocol.
After a 20-min equilibration period, the pancreas (n = 7) was perfused with the basal perfusion medium (3 mmol/l glucose: minutes 0–40) and thereafter at 11 mmol/l (minutes 41–80), for 40 min each. During both periods, 1 nmol/l GLP-1 was co-administered for 10 min (minutes 16–25 and 56–65, respectively), allowing hormone secretion to stabilize before and after application. After cessation of the 11 mmol/l period, the pancreas was perfused for 10 min with the basal perfusion medium at 3 mmol/l glucose (minutes 81–90), during which time endocrine secretion returned to basal levels. Then the same protocol was repeated, i.e., perfusion with 3 and 11 mmol/l glucose-containing medium for 40 min each (minutes 91–130 and 131–170, respectively) and administration of 1 nmol/l GLP-1 for 10 min each (minutes 106–115 and 146–155, respectively), with the sole exception that tolbutamide at a final concentration of 185 μmol/l (50 mg/l) in the perfusion medium was constantly co-infused from minute 91 until the end of perfusion at minute 180. In control experiments (n = 6), the same protocol was followed, except that no GLP-1 was added.
Hormonal analysis.
Pancreatic insulin, glucagon, and somatostatin concentrations in the venous effluent were analyzed by radioimmunoassay. Insulin concentrations were determined using guinea pig antiserum raised against porcine insulin (2006-3), which cross-reacts strongly with both rat insulin I and II and human 125I-labeled insulin labeled in position A14. A mixture (2:1) of rat insulin I and II was used as standard (Novo Nordisk) (19). Glucagon immunoreactivity was measured with the COOH-terminally directed antiserum 4305, which mainly detects glucagon of pancreatic origin, and highly purified porcine glucagon as standard (20). Somatostatin immunoreactivity was determined using rabbit antiserum 1758, raised against synthetic cyclic somatostatin (SS), recognizing both SS-14 and SS-28 (21).
Calculations and statistical analysis.
Results are expressed as means ± SE in picomoles per minute for an indicated number of experiments. Statistical evaluation consisted of two-way ANOVA and two-tailed t test for unpaired data followed by Bonferroni correction for multiple comparisons, as well as Spearman’s rank correlation of mean hormone outputs of corresponding prestimulatory (the 5-min period preceding the initiation of the infusion), stimulatory (the 5-min period, during which maximal hormone response was observed), and stable poststimulatory periods. In addition, 1-min glucagon and somatostatin outputs of the first 90 min were correlated in each individual perfusion experiment in which GLP-1 was given (n = 7). All analyses were performed using StatSoft (2004) (Statistica, version 7). Values of P < 0.05 were considered significant.
RESULTS
Insulin.
At 3 mmol/l glucose, GLP-1 alone did not stimulate insulin secretion (2.0 ± 0.6 vs. 1.4 ± 0.5 pmol/min for basal secretion, mean ± SE) (Fig. 1, bold line). In contrast, in control experiments without GLP-1, tolbutamide alone caused a significant increase in insulin secretion with a rapid peak (46.0 ± 12.9 pmol/min vs. basal secretion; P < 0.0001) followed by a sustained phase at a significantly lower level (11.3 ± 3.8 pmol/min vs. basal secretion; NS). In GLP-1 experiments, the insulin response to tolbutamide alone was significantly higher than in the corresponding time interval in control experiments (88.4 ± 10.3 vs. 46.0 ± 12.9 pmol/min; P = 0.024), even though the mean hormone outputs during the 5-min periods immediately preceding the stimulation with tolbutamide (3 mmol/l glucose only) did not differ significantly from each other.
However, when GLP-1 and tolbutamide were given simultaneously at 3 mmol/l glucose, the insulin response increased to 43.7 ± 6.2 pmol/min, exceeding the sum of the corresponding individual hormone responses to GLP-1 alone at 3 mmol/l glucose (2.0 ± 0.6 pmol/min; P = 0.012) and tolbutamide alone at 3 mmol/l glucose (11.3 ± 3.8 pmol/min; P = 0.009) by a factor of 3.3.
Perfusion with 11 mmol/l glucose alone induced a biphasic insulin secretion, the second phase of which was significantly enhanced by administration of GLP-1 (115.4 ± 14.1 vs. 30.1 ± 7.6 pmol/min, P = 0.001). During tolbutamide infusion, 11 mmol/l glucose also caused first- and second-phase insulin secretion, and when GLP-1 and tolbutamide were co-administered, the insulin output increased to 141.7 ± 10.3 pmol/min, compared with tolbutamide alone at 11 mmol/l glucose (42.5 ± 7.3 pmol/min; P < 0.0001) and GLP-1 alone at 11 mmol/l glucose (115.4 ± 14.1 pmol/min; NS). In GLP-1 experiments, the immediate insulin response to tolbutamide alone at 11 mmol/l was significantly higher than in control experiments (49.5 ± 4.0 vs. 22.7 ± 6.1 pmol/min; P = 0.006).
Glucagon.
At 3 mmol/l glucose, glucagon secretion was inhibited by GLP-1 from 253.4 ± 12.3 fmol/min (basal secretion) to 180.5 ± 14.0 fmol/min (P = 0.031) (Fig. 2, bold line). After cessation of the GLP-1 infusion, glucagon output recovered to 367.7 ± 31.4 fmol/min, thereby exceeding baseline values significantly (P = 0.001 vs. basal secretion).
Perfusion with 11 mmol/l glucose suppressed glucagon secretion strongly in comparison to basal secretion at 3 mmol/l glucose in both GLP-1 and control experiments (GLP-1: 253.4 ± 12.3 fmol/min [basal secretion] vs. 102.7 ± 11.2 fmol/min [11 mmol/l], P < 0.0001; control: 307.9 ± 79.2 fmol/min [basal secretion] vs. 105.1 ± 10.0 fmol/min [11 mmol/l], P = 0.006). At 11 mmol/l glucose, GLP-1 caused a small but statistically significant further attenuation of glucagon secretion from 102.7 ± 11.2 to 87.1 ± 8.6 fmol/min (P = 0.023).
During the short period of reperfusion with 3 mmol/l glucose alone and during infusion with tolbutamide thereafter, glucagon secretion slightly increased (NS). However, during tolbutamide infusion, glucagon output did not recover and remained at a significantly lower level compared with basal secretion (GLP-1 experiments: 156.7 ± 28.6, P = 0.010; control: 146.9 ± 19.1 fmol/min, P = 0.033; both versus basal secretion at 3 mmol/l glucose). Addition of GLP-1 during tolbutamide infusion did not induce significant changes in glucagon secretion in comparison to control experiments at either 3 or 11 mmol/l glucose.
Somatostatin.
At 3 and 11 mmol/l glucose, GLP-1 increased somatostatin secretion (Fig. 3, bold line) compared with both prestimulatory output (3 mmol/l: 39.4 ± 8.0 vs. 24.7 ± 5.8 fmol/min, P = 0.041; 11 mmol/l: 71.0 ± 12.2 vs. 35.8 ± 6.0 fmol/min, P = 0.019) and secretion in control experiments, in which GLP-1 was omitted (3 mmol/l: 12.9 ± 4.0 fmol/min, P = 0.068; 11 mmol/l: 24.9 ± 3.7 fmol/min, P = 0.026). The increase in glucose concentration itself induced a further statistically significant rise in secretion from 19.3 ± 2.9 to 35.8 ± 6.8 fmol/min (P = 0.005, GLP-1 experiments) and 10.1 ± 2.6 to 22.1 ± 3.6 fmol/min (P = 0.036, control).
Infusion of tolbutamide alone at 3 mmol/l provoked a strong rise in somatostatin output to 82.1 ± 14.5 fmol/min (P = 0.004 vs. basal secretion) in GLP-1 experiments and 71.2 ± 15.7 fmol/min (P = 0.006 vs. basal secretion) in control experiments, respectively. Co-infusion of GLP-1 enhanced somatostatin secretion further to 115.1 ± 22.4 fmol/min at 3 mmol/l (P = 0.002 vs. prestimulatory output; P = 0.073 vs. control) and 132.5 ± 26.2 fmol/min at 11 mmol/l glucose (P = 0.007 vs. prestimulatory output; P = 0.039 vs. control).
Analysis of correlation.
Analysis of 5-min mean hormone outputs showed that glucagon secretion was negatively correlated with both somatostatin (Spearman rank correlation coefficient rs = −0.511; P < 0.001) and insulin secretion (rs = −0.710; P < 0.001), whereas insulin and somatostatin secretion showed a positive correlation (rs = 0.657; P < 0.001) (Table 1). Analysis of 1-min glucagon and somatostatin outputs demonstrated significant inverse correlations for each of the seven experiments (mean rs = −0.606 ± 0.08).
DISCUSSION
In this study, the influence of the sulfonylurea tolbutamide on the glucose dependence of the GLP-1–mediated effects on insulin, glucagon, and somatostatin secretion in the perfused rat pancreas was examined. The most important finding was that simultaneous infusion of tolbutamide enabled GLP-1 to stimulate insulin secretion, even at 3 mmol/l glucose, where GLP-1 alone had no effect. Consistent with our results, GLP-1 has been shown to be ineffective in stimulating insulin secretion below a threshold glucose level of 4.3 mmol/l in humans (6).
Contact of the β-cell with adequate glucose levels (>7–8 mmol/l) elicits a sequence of metabolic events resulting in ATP generation, closure of KATP channels, membrane depolarization, and Ca2+-dependent exocytosis of insulin-containing granules (3). GLP-1 directly modifies the activity of KATP and L-type Ca2+ channels, mobilizes extra- and intracellular Ca2+ sources, and recruits and primes insulin-containing granules, thereby enforcing the overall insulin output (3,12,13,22–25). However, despite the many effects GLP-1 exerts in favor of promoting insulin secretion, it is incapable of triggering insulin secretion itself. This might be because GLP-1 alone cannot accomplish sufficient inhibition of KATP channels (>90%), which is essential for the initiation of membrane depolarization and other downstream steps in the insulin secretion signaling pathway (3,22,26). Instead, high glucose levels or the concomitance of GLP-1 and intermediate glucose levels are necessary.
By applying the KATP channel-inhibitor tolbutamide in a dose of 185 μmol/l (50 mg/l), which is within the therapeutic range in humans, we bypassed the precondition of elevated glucose levels, and showed GLP-1-induced insulin secretion at a glucose concentration of 3 mmol/l (17,27). These results are in line with clinical trials, in which the incidence of mild to moderate hypoglycemia was greatly increased in diabetic patients receiving combination therapy of the GLP-1 mimetic exendin-4 and sulfonylurea (8–11). Since sulfonylurea act even at very low glucose concentrations, their use is often compromised by the risk of hypoglycemia (28). In view of the strict glucose dependence of GLP-1, it is tempting to speculate that the higher occurrence of adverse events in this group of patients therefore is solely due to the intake of sulfonylurea compounds.
In the present study, however, at a glucose concentration of 3 mmol/l, the insulin secretion induced by simultaneous infusion of GLP-1 and tolbutamide exceeded the sum of the corresponding individual insulin responses by a factor of 3.3. The insulin response to 11 mmol/l glucose was not significantly different whether tolbutamide was absent or present and included a first-phase response, perhaps because glucose was still capable of closing further KATP channels incompletely blocked by tolbutamide. The ability of GLP-1 to further enhance insulin secretion during perfusion with 11 mmol/l glucose and tolbutamide (although not significant) might be the result of a synergistic interaction between GLP-1 and tolbutamide (see below). Addition of GLP-1 during ongoing tolbutamide infusion at 3 mmol/l glucose significantly stimulated insulin secretion, demonstrating uncoupling of GLP-1 from its glucose dependence. These results extend previous observations by Gutniak et al. (29), who showed a reinforcement of insulin secretion by the combination of GLP-1 and the sulfonylurea glibenclamide in diabetic patients with secondary failure to sulfonylurea and in the perfused rat pancreas at a glucose concentration of 3.3 mmol/l. Our findings are also consistent with the observation that both dibutyryl-cAMP and GLP-1 administered together with tolbutamide increased the cytosolic free Ca2+ concentration at 2.8 mmol/l glucose in single rat pancreatic β-cells (30).
Based on our experiments, we can deduce that co-infusion of tolbutamide, and thereby most likely inhibition of KATP channels, does play a role in the glucose dependence of GLP-1 or at least its initiation, since the increment in insulin secretion at 3 mmol/l glucose observed in our experiments could only be evoked in the presence of tolbutamide. However, the precise mechanism by which GLP-1 and tolbutamide interact to induce this potentiation and mobilize this additional insulin secretion cannot be determined from the present study. One possible explanation might be the recruitment and inhibition of remaining KATP channels, since we cannot determine exactly to which extent these were closed by the tolbutamide dose (185 μmol/l) applied in our experiments, which was primarily chosen to reflect therapeutic concentrations in humans. Using excised inside-out patches, Gribble et al. (31) demonstrated that the mean tolbutamide concentration at which inhibition of wild-type KATP channels is half-maximal is 2 μmol/l, while in whole-cell preparations, 100 μmol/l tolbutamide decreased the K+ conductance by 90–95% (32). Another explanation could be GLP-1’s direct effect on secretory granules and facilitation of their exocytosis, which in response to the rise in intracellular calcium concentration by tolbutamide will be amplified (13). Thus, further studies to explore the dose-response relationships and mechanism of the synergistic interaction between GLP-1 and tolbutamide at low glucose concentrations are warranted.
Because hyperglucagonemia contributes to the development and progression of type 2 diabetes, substantial interest has been focused on the mechanisms underlying the regulation of glucagon secretion from pancreatic α-cells (33). Interestingly, KATP channels corresponding to those expressed in pancreatic rat β-cells seem to be involved in the glucose-dependent stimulus-secretion coupling in α-cells (34–36). However, by reason of specific voltage-dependent Na+ channels, which become inactivated when the membrane potential becomes too positive, high glucose concentrations with subsequent KATP channel closure and membrane depolarization will not lead to exocytosis and hormone secretion as in β-cells (36,37). At low glucose, conversely, KATP channels are open and cause a weak hyperpolarization with subsequent activation of Na+- and T-type Ca2+ channels, Ca2+ influx, and glucagon release (36,37). These findings are in line with our study, in which glucagon secretion was significantly suppressed by perfusion with 11 mmol/l glucose alone. However, secretion recovered only slightly during the subsequent perfusion with 3 mmol/l glucose alone, which might be due to the short perfusion period of 10 min.
While it is well established that hyperglycemia suppresses glucagon secretion strongly, the effect of sulfonylurea on glucagon secretion is controversial and reported to be unaffected (17), stimulated (38), inhibited (39), and influenced differently depending on the prevailing glucose and drug concentration and presence of paracrine effectors (38,40,41). We observed that during infusion with tolbutamide alone, glucagon secretion did not rise in the presence of 3 mmol/l glucose. Perhaps, because of tolbutamide-induced inhibition of KATP channels, a condition resembling hyperglycemia is established, which according to the above model, will lead to inactivation of voltage-gated Na+ and Ca2+ channels with subsequent suppression of electrical activity and glucagon release. This hypothesis is supported by findings by Göpel et al. (36), who, after addition of tolbutamide, observed a strong depolarization, but only small and irregular oscillations in membrane potential in patch-clamp experiments performed on single mouse α-cells.
GLP-1 potently inhibits glucagon secretion, presumably by protein kinase A–dependent inhibition of KATP channel activity (2,37). In our study, GLP-1 reduced glucagon secretion significantly at 3 mmol/l glucose, whereas only a slight effect was seen at 11 mmol/l glucose, at which point glucagon levels were already strongly suppressed by hyperglycemia. During tolbutamide infusion, GLP-1 did not induce significant differences in glucagon secretion compared with control experiments. Possibly, KATP channel activity was already potently inhibited by tolbutamide alone at 3 mmol/l glucose, precluding a further inhibitory effect of GLP-1.
However, the model described above does not take into account the influence of paracrine factors on glucagon regulation. Somatostatin, secreted from pancreatic δ-cells, potently inhibits glucagon secretion, whereas glucose and GLP-1 stimulate somatostatin release (4,42–45). It seems possible that somatostatin acts as an important paracrine regulator of glucagon secretion and that GLP-1, in addition, mediates its glucagonostatic effects on α-cells indirectly via somatostatin (46). This hypothesis is supported by the finding that somatostatin inhibits Ca2+-induced exocytosis in rat pancreatic α-cells (47). In our study, at 3 mmol/l glucose, only a small amount of somatostatin was released and thus did not inhibit glucagon secretion, which in contrast was enhanced. However, infusion of GLP-1 induced a significant increase in somatostatin release accompanied by an inhibition of glucagon secretion, which did not occur in control experiments. The rise in glucose concentration from 3 to 11 mmol/l also augmented somatostatin output and was paralleled by a sharp decline in glucagon secretion. At 11 mmol/l glucose, GLP-1 induced a pronounced increase in somatostatin output, resulting in a small, but significant, reduction in glucagon secretion. As mentioned above, the minor inhibitory effect of GLP-1, despite the potent increase in somatostatin output, might be due to the already strong suppression of glucagon secretion. The same might apply for the sulfonylurea-induced restraint of glucagon secretion, since administration of tolbutamide significantly increased somatostatin secretion, as reported by others, while at the same time, a counteractive rise in glucagon secretion failed to appear in spite of the low perfusate glucose concentration (45).
Accordingly, in our study, a strong negative correlation between somatostatin and glucagon secretion was observed both when 1-min hormone outputs were analyzed in individual perfusion experiments and when the 5-min mean hormone outputs for all experiments were correlated. In addition, glucagon and insulin secretion were negatively correlated and somatostatin and insulin secretion positively correlated, indicating that insulin is also involved in the complex intra-islet regulation of hormone secretion as postulated by others (5).
Regarding stimulus-secretion coupling in δ-cells, inhibition of KATP channels with subsequent increases in intracellular Ca2+ levels via voltage-gated Ca2+ channels seems to be involved (48,49). In accordance with the findings in β-cells, tolbutamide, like a glucose stimulus, might induce KATP channel closure and membrane depolarization, leading to somatostatin secretion, as observed in our study (49). At present, few data exist elucidating the mechanisms by which GLP-1 stimulates somatostatin secretion. It is tempting to speculate that GLP-1 might affect signaling in a similar manner as in β-cells, by enhancing KATP channel closure and Ca2+ influx. Interestingly, in contrast to the glucose dependence of its insulinotropic actions, GLP-1 stimulates somatostatin secretion even at low glucose levels and does not depend on concomitant high glucose levels or tolbutamide as a prerequisite to trigger somatostatin release. Maybe the existence of specific voltage-dependent Na+ channels, which have also been demonstrated in α-cells, contributes to this differential behavior (49). Further investigations of the GLP-1–mediated stimulation of somatostatin are needed, especially to elucidate the interplay of somatostatin and GLP-1 in the regulation of glucagon secretion.
In conclusion, we demonstrated uncoupling of the insulinotropic action of GLP-1 from its glucose dependence by administration of the KATP channel blocker tolbutamide, thereby suggesting a role of KATP channels as a link between environmental glucose concentration and the β-cell’s secretory response. However, from our experiments, we cannot determine precisely at which cellular/molecular level GLP-1 acts to potentiate insulin secretion in synergism with tolbutamide at this low glucose concentration. Further studies are required to investigate the underlying mechanism of this uncoupling, which might explain the high occurrence of hypoglycemia in diabetic patients receiving combination therapy of sulfonylurea and GLP-1.
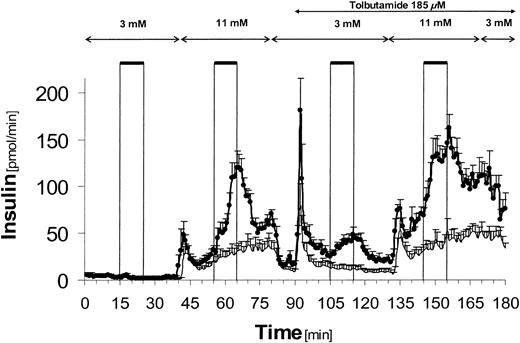
Insulin secretion from the perfused rat pancreas. •, Experiments with addition of GLP-1 (1 nmol/l) (n = 7). Solid line, control experiments without addition of GLP-1 (n = 6). The arrows indicate infusion of different glucose concentrations and co-infusion of tolbutamide (185 μmol/l). The bars indicate when GLP-1 (1 nmol/l) was infused for 10 min in GLP-1 experiments. Each data point represents the mean of six to seven experiments ± SE.
Insulin secretion from the perfused rat pancreas. •, Experiments with addition of GLP-1 (1 nmol/l) (n = 7). Solid line, control experiments without addition of GLP-1 (n = 6). The arrows indicate infusion of different glucose concentrations and co-infusion of tolbutamide (185 μmol/l). The bars indicate when GLP-1 (1 nmol/l) was infused for 10 min in GLP-1 experiments. Each data point represents the mean of six to seven experiments ± SE.
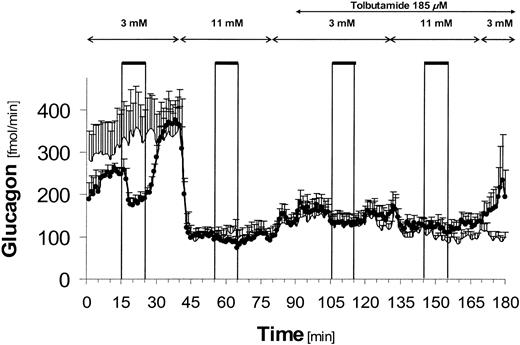
Glucagon secretion from the perfused rat pancreas. •, Experiments with addition of GLP-1 (1 nmol/l) (n = 7). Solid line, control experiments without addition of GLP-1 (n = 6). The arrows indicate infusion of different glucose concentrations and co-infusion of tolbutamide (185 μmol/l). The bars indicate when GLP-1 (1 nmol/l) was infused for 10 min in GLP-1 experiments. Each data point represents the mean of six to seven experiments ± SE.
Glucagon secretion from the perfused rat pancreas. •, Experiments with addition of GLP-1 (1 nmol/l) (n = 7). Solid line, control experiments without addition of GLP-1 (n = 6). The arrows indicate infusion of different glucose concentrations and co-infusion of tolbutamide (185 μmol/l). The bars indicate when GLP-1 (1 nmol/l) was infused for 10 min in GLP-1 experiments. Each data point represents the mean of six to seven experiments ± SE.

Somatostatin (SS) secretion from the perfused rat pancreas. •, Experiments with addition of GLP-1 (1 nmol/l) (n = 7). Solid line, control experiments without addition of GLP-1 (n = 6). The arrows indicate infusion of different glucose concentrations and co-infusion of tolbutamide (185 μmol/l). The bars indicate when GLP-1 (1 nmol/l) was infused for 10 min in GLP-1 experiments. Each data point represents the mean of six to seven experiments ± SE.
Somatostatin (SS) secretion from the perfused rat pancreas. •, Experiments with addition of GLP-1 (1 nmol/l) (n = 7). Solid line, control experiments without addition of GLP-1 (n = 6). The arrows indicate infusion of different glucose concentrations and co-infusion of tolbutamide (185 μmol/l). The bars indicate when GLP-1 (1 nmol/l) was infused for 10 min in GLP-1 experiments. Each data point represents the mean of six to seven experiments ± SE.
Correlation between glucagon, somatostatin, and insulin secretion
| Glucagon | Somatostatin | Insulin | |
|---|---|---|---|
| Glucagon | — | −0.511* | −0.710* |
| Somatostatin | −0.511* | — | 0.657* |
| Insulin | −0.710* | 0.657* | — |
| Glucagon | Somatostatin | Insulin | |
|---|---|---|---|
| Glucagon | — | −0.511* | −0.710* |
| Somatostatin | −0.511* | — | 0.657* |
| Insulin | −0.710* | 0.657* | — |
Data are Spearman rank correlation coefficients rs calculated from 5-min mean hormone outputs of stable prestimulatory, poststimulatory, and stimulatory periods of the first 90 min of all experiments.
P < 0.001.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Article Information
This work was supported by the German Research Foundation (to J.d.H., HE 3639/1-1), the Danish Medical Research Council, and the European Foundation for the Study of Diabetes.